Supramolecular Chemistry of Fullerenes
Supramolecular Chemistry
1.-Complexation of Fullerenes
The convex geometry of fullerenes together with its large π surface makes them particularly well-suited for its molecular recognition by concave conjugated fragments.1
As a result, during the last years, we have mainly worked with π-extended derivatives of tetrathiafulvalene (exTTF) as novel receptors for fullerenes given the shape complementarity of its concave aromatic. However, in spite of theoretical calculations predicting binding energies of around 7 kcal·mol-1 in the gas phase, attempts to combine both moieties through simply π-π interactions remained elusive at first. Thus, a number of different strategies have been developed in order to get over these difficulties (see figure). Other receptors such as porphyrins and TCAQ have also been developed in our lab.
Increasing the electron donor character and the π surface

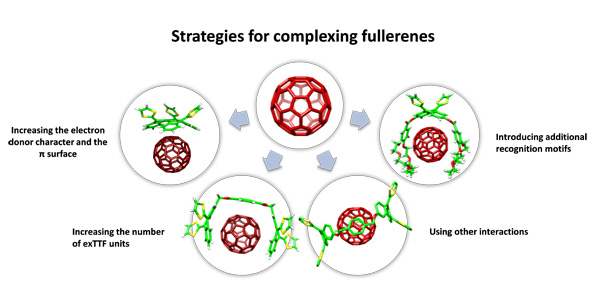
Trux-TTF·hemifullerene complex. This paper reports the first time that a fullerene fragment has been found to mimic the behavior of C60 as an electron acceptor.
Based on the same concave-convex complementarity principle, and with the aim to construct an electron-donor fragment that would: 1) show good electron-donor properties, 2) absorb light efficiently, preferably in the visible region and 3) self-assemble in a controlled fashion, we started working with a new family of exTTF derivatives, the truxene-TTFs which have a third dithiole ring in a truxene core offering additional benzene rings.
To accommodate the dithioles, the truxene moiety breaks down its planar structure and adopts an all-cis sphere-like geometry with the three dithiole rings protruding outside, as shown in its X-ray crystal structure. The concave bowl-shape configuration adopted by the truxene core perfectly mirrors the convex surface of fullerenes, suggesting that van der Waals and concave-convex interactions between them should be maximized. Indeed, the association of trux-TTF and fullerenes in solution was investigated by 1H NMR titrations with C60 and C70 as guests affording binding constants of Log Ka = 3.1 and 3.9 for C60 and C70, respectively.2
Very recently, we have also discovered that trux-TTFs are able to complex as well hemifullerenes forming heteromolecular dimers in solution with binding constants on the order of 103 M-1.3
Increasing the number of exTTF units

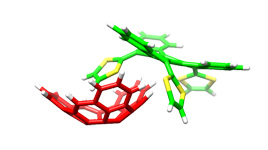
exTTF based supramolecular polymer. This paper reports one of the few examples of supramolecular polymers in which π-π aromatic interactions are the chief driving force for polymerization.
The first exTTF based receptor was initially based on simple tweezers-like designs;4 and showed binding constants in the order of Log Ka= 3.5 in chlorobenzene at room temperature.5 Based on this strategy, we have been able to construct more complex self-assembled donor-acceptor nanostructures such as linear6 and superbranched7 supramolecular polymers or large covalent dendrimers up to 4th generations capable of associating several units of C60.8 The analogous all-acceptor linear and hyperbranched supramolecular oligomers have also been constructed by substituting the exTTF units with the geometrically equivalent tetracyanoanthraquinodimethane (TACQ).9
Combining the cyclotriveratrilene (CTV) scaffold together with three units of exTTF covalently connected led to a receptor with very large affinities of Log Ka= 5.3 towards C60 and Log Ka= 6.3 towards C70 (all binding constants measured in PhCl, at 298 K).10
Using other interactions
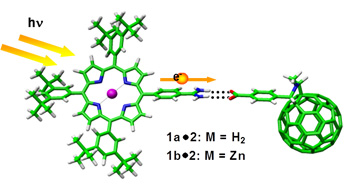
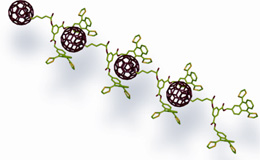
C60 -porphyrin dimer stabilized by a two-point amidinium-carboxylate. This paper reports one of the scarce examples of electron transfer through a hydrogen bond.
Pioneering work carried out by Sessler13 and Therien14 demonstrated that the electronic communication through hydrogen-bonding interfaces is more efficient than in comparable σ- or π-bonding networks.
Following this seminal work, we have investigated a set of non-covalently associated C60-porphyrin ensembles interfaced by a two-point amidinium-carboxylate pair that facilitates an efficient charge separation process to afford microsecond-lived P•+-C60•- radical pairs. In solvents that do not interfere significantly with either the electrostatic or the hydrogen bonding interactions binding constants as high as Log Ka= 7.3 were observed.11 Exceptionally strong electronic couplings stem from this self-assembly, which in turn facilitates faster, more efficient and longer-lived formation of radical ion pair states when compared to similar covalent C60 conjugates. Most importantly, such remarkable radical ion pair lifetimes outperform previously reported ensembles based on non amidinium-carboxylate binding motif or non fullerene electron acceptors by several orders of magnitude.
Recently, we have published a comprehensive investigation of the supramolecular wire behaviour of a new set of hybrid covalent/supramolecular porphyrin-fullerene structures, in which hydrogen-bond interactions and p-phenylene oligomers of different length act as highly efficient molecular wires exhibiting a remarkably low attenuation factor (β= 0.07 ± 0.01 Å-1).15
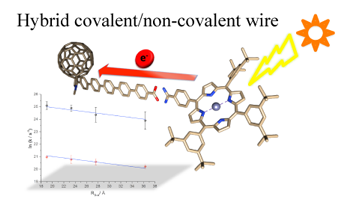
A hydrogen bond pseudo-rotaxane with a fullerene-based secondary ammonium salt was also obtained via hydrogen bond. However, the stability of this complex was very low, probably due to the big and hindered cavity.12 To overcome this limitation, a series of exTTF-based secondary ammonium salts were complexed with a smaller DB[24]C8fullerene obtaining higher Ka values.13
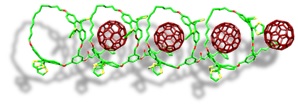
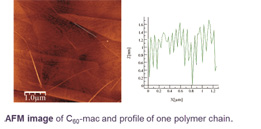
exTTF based supramolecular polymer formed by macrocycles. Mass spectroscopy reports the presence of polymers of up to 43 units forming fibers visible by AFM.
Finally, we have also developed some of the best receptors for fullerenes reported in the literature by exploiting the concept of macrocycle effect in a series of exTTF based tweezers with Log Ka= 6.5 towards C60 in PhCL and 7.5 in PhCN at room temperature.14 This approach enabled us to develop a new supramolecular polymer much more stable than its acyclic analogous.16
Introducing additional recognition motifs
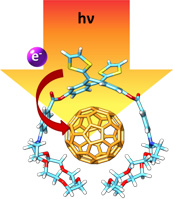
exTTF·(crown ether)2 tweezer. The use of crown ethers as an additional recognition motif increased the overall stability of the complex by more than 2 orders of magnitude.
A wise combination of different supramolecular interactions can lead to a dramatic increase of the overall stability of the complex due to cooperative effects.
In this regard, we have successfully combined together hydrogen bonds and exTTF-fullerene interaction to construct extremely stable (Log Ka = 6.2) exTTF-C60 dyads. To do so, we synthesized an exTTF derivative bearing a crown ether moiety and a Bingel-type C60 adduct featuring an ammonium salt. The combination of both interactions produced an increase of more than three orders of magnitude of the complex in solution. Furthermore, we have demonstrated that upon light irradiation, these supramolecular species yield a charge separated state with a lifetime of 9.3 ps.17
Very recently we have studied the potentiality of crown ethers as recognition motifs offering cooperative π- π and n- π interactions. With this purpose, we have decorated a single exTTF unit with two crown ether moieties, obtaining log Ka= 7.0 in PhCN at room temperature. This is the first example of a single exTTF unit able to complex fullerenes.18
2.- Self-Assembly
The construction of self-assembling and replicating structures that bear photonic and/or electronic active units at the nanometric scale constitutes one of the biggest challenges in contemporary science. In the bottom-up approach, control over the self-organizing constituents of unprecedented one-, two-, and three-dimensional nanostructured materials at different length scales is of primary interest for the fine tuning of electronic and optical properties.
With this in mind, and inspired by the manifold possibilities of “curved” scaffolding π-exTTF species, we have explored the creation of novel photo- and electroactive 3D nanoarchitectures.
We have investigated a new type of noncovalent assembly, in which the nonplanar and redox-active π-exTTF units associate through π–π interactions, leading to their organization into fibers. This study has shown for the first time that butterfly-shaped π-exTTFs can facilitate the efficient organization of new 3D materials at the nanometer scale. The different nanostructures and morphologies resulting from the primary, secondary, tertiary, and quaternary architectures have been confirmed by a variety of complementary techniques (XRD, AFM, TEM, and SEM). Photophysical measurements confirmed that the quaternary structure enables remarkable stabilization of the photogenerated radical ion pair, which has a lifetime in the millisecond range in the supramolecular assembly.19-20
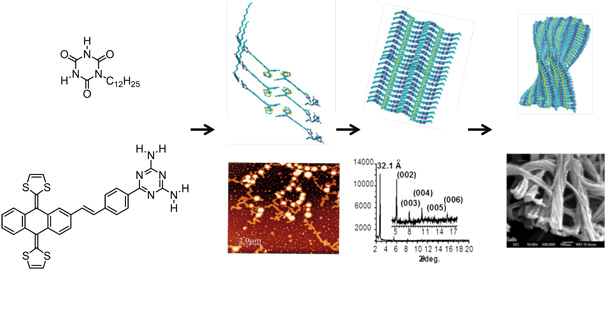
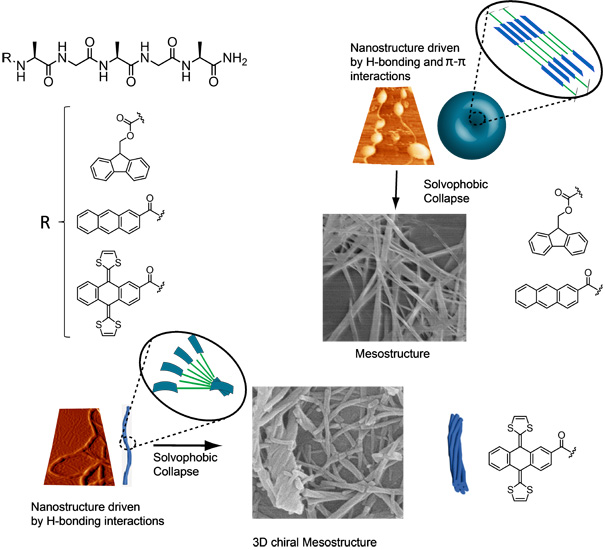
On the other hand, we have designed and synthesized a new assembling based on an aromatic peptide amphiphilic system with alanyl-glycyl-alanyl-glycyl-alanine (AGAGA) pentapeptide sequences that are linked to exTTF. The presence of short peptide sequences eases the formation of chiral β sheet as secondary structure. The introduction of bulky exTTF in this system, with its concave central core near the β sheet forming peptide sequences, induces torsional strains in the secondary H-bonded nanostructure. The consequences are twofold: firstly, a maximum intermolecular H bonding within the peptide backbone is preserved. Secondly, mutual interactions between exTTFs are avoided. In turn, 1D helical nanostructures are realized at the nanoscale driven exclusively by H-bonding interactions. The latter seem to be susceptible to a subsequent self-assembly process at mesoscopic length scales. In fact, π–π interacting exTTFs merge under solvophobic conditions into 3D highly ordered hierarchical architectures with helical features.
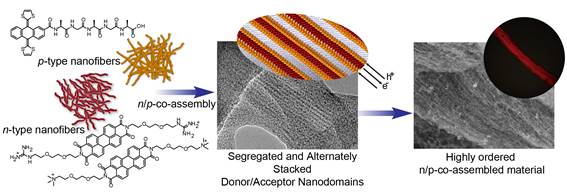
The precise control over the organization of photoactive components at the nanoscale is one of the main challenges for the generation of new and sophisticated macroscopically ordered materials with enhanced properties. Herein, we have developed two approach for the construction of highly ordered electroactive materials: i) Inspired by the previously results obtained in the formation of n-type electroactive fiber based on sequence peptides covalent linked to p-extended tetrathiofulvalene, we have developed the construction of highly ordered donor/acceptor functional material, using the principle of ionic self-assembly. An electron donor π-extended tetrathiafulvalene and an electron acceptor perylene-bisimide were self-organized separately obtaining n- and p-nanofibers at the same scale. These complementary n- and p-nanofibers are endowed with ionic groups with opposite charges on their surfaces. The synergic interactions establish periodic alignments between both nanofibers resulting in a material with segregated and alternately stacked donor/acceptor nanodomains. Photoconductivity measurements show values for these n/p-co-assembled materials up to 0.8 cm2V-1s-1, confirming the effectiveness in the design of these hetero-junction structures. This easy methodology offers great possibilities to achieve highly ordered n/p-materials for potential applications in different areas such as optoelectonics and photovoltaic.21
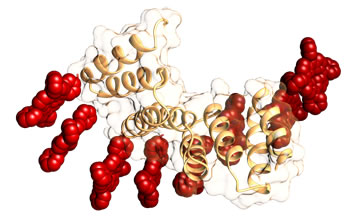
ii) An second approach is based on the use of protein-based building blocks for the arrangement of photo and electroactive porphyrin derivatives. We used a designed repeat protein scaffold with demonstrated unique features that allow for the control of their structure, functionality, and assembly. Our designed domains act as exact biomolecular templates to organize porphyrin molecules at the required distance. The hybrid conjugates retain the structure and assembly properties of the protein scaffold and display the spectroscopic features of orderly aggregated porphyrins along the protein structure. Moreover, we achieved a solid ordered bio-organic hybrid thin film with anisotropic photoconductivity.

References:
1.(a) E. M. Pérez, N. Martín, Chem. Soc. Rev. 2008, 37, 1512-1519; (b) Supramolecular Chemistry of Fullerenes and Carbon Nanotubes, (Ed. N. Martín, J.-F. Nierengarten), Wiley-VCH, Weinheim, 2012.
2. E. M. Pérez, M. Sierra, L. Sánchez, M. R. Torres, R. Viruela, P. M. Viruela, E. Ortí and N. Martín, Angew. Chem. Int. Ed. 2007, 46, 1847-1851.
3. M. Gallego, J. Calbo, J. Aragó, R. M. Krick Calderon, F. H. Liquido, T. Iwamoto, A. K. Greene, E. A. Jackson, E. M. Pérez, E. Ortí, D. M. Guldi, L. T. Scott, N. Martín, Angew. Chem. Int. Ed.,2014, 53, 2170-2175.
4. E. M. Pérez, N. Martín, Pure Appl. Chem. 2010, 82, 523-533.
5. (a) E. M. Pérez, L. Sánchez, G. Fernández and N. Martín, J. Am. Chem. Soc. 2006, 128, 7172-7173; (b) E. M. Pérez, A. L. Capodilupo, G. Fernández, L. Sánchez, P. M. Viruela, R. Viruela, E. Ortí, M. Bietti and N. Martín, Chem. Commun. 2008, 4567-4569; (c) S. S. Gayathri, M. Wielopolski, E. M. Pérez, G. Fernández, L. Sánchez, R. Viruela, E. Ortí, D. M. Guldi and N. Martín, Angew. Chem. Int. Ed. 2009, 48, 815-819.
6. G. Fernández, E. M. Pérez, L. Sánchez and N. Martín, Angew. Chem. Int. Ed. 2008, 47, 1094-1097.
7. G. Fernández, E. M. Pérez, L. Sánchez and N. Martín, J. Am. Chem. Soc. 2008, 130, 2410-2411.
8. G. Fernández, L. Sánchez, E. M. Pérez, N. Martín, J. Am. Chem. Soc., 2008, 130, 10674-10683.
9. J. Santos, E. M. Pérez, B. M. Illescas, N. Martín, Chem. Asian. J., 2011, 6, 1848-1853.
10. E. Huerta, H. Isla, E. M. Pérez, C. Bo, N. Martín and J. de Mendoza, J. Am. Chem. Soc. 2010, 132, 5351.
11. L. Sánchez, M. Sierra, N. Martín, A. J. Myles, T. J. Dale, J. Rebek, W. Seitz and D. M. Guldi, Angew. Chem. Int. Ed. 2006, 45, 4637-4641.
12. M. C. Díaz, B. M. Illescas, N. Martín, J. F. Stoddart, M. A. Canales, J. Jiménez-Barbero, G. Sarova and D. M. Guldi, Tetrahedron 2006, 62, 1998-2002.
13. B. M. Illescas, J. Santos, M. C. Díaz, N. Martín, C. M. Atienza and D. M. Guldi, Eur. J. Org. Chem. 2007, 2007, 5027-5037.
14. (a) H. Isla, M. Gallego, E. M. Pérez, R. Viruela, E. Ortí, N. Martín, J. Am. Chem. Soc., 2010, 132, 1772-1773; (b) D. Canevet, M. Gallego, H. Isla, A. de Juan, E. M. Pérez and N. Martín, J. Am. Chem. Soc. 2011, 133, 3184-3190.
15. Angew. Chem. Int. Ed. 2016, DOI: 10.1002/anie.201608973.
16. Angewandte Submitted, (in press)
17. J. Santos, B. Grimm, B. M. Illescas, D. M. Guldi and N. Martín, Chem. Commun. 2008, 5993-5995.
18. B. Grimm, J. Santos, B. M. Illescas, A. Muñoz, D. M. Guldi and N. Martín, J. Am. Chem. Soc. 2010, 132, 17387-17389.
19. J. L. López, C. Atienza, W. Seitz, D. M. Guldi, N. Martín, Angew. Chem. Int. Ed., 2010, 49, 9876-9880.
20. J. L. López, C. Atienza, A. Insuasty, J. López-Andarias, C. Romero-Nieto, D. M. Guldi, N. Martín, Angew. Chem. Int. Ed., 2012, 51, 3857-3861.
21. J. Am. Chem. Soc. 2015, 137, 893
22 Chem. Sci. 2016, 7, 4842-4847