Molecular Electronics
Molecular electronics
The rational design of molecular-sized materials for electronic and photonic applications is currently a topic of great interest. In particular, the preparation and integration of multifunctional molecules into architectures of higher order -the so called bottom-up approach- is the basis for the realization of molecular-scale electronic.
A molecular wire consists of a molecular unit connected to two continuum reservoirs of electrons where the wire provides a pathway for the transport of electrons from one reservoir to another. Hereby, p-conjugation emerges as a particular crucial factor to realize i) small attenuation factors (i.e., electron conducting over large distances), ii) good contacts with the termini (i.e., electron donors and electron acceptors) and iii) good orbital mixing (i. e., with donor and acceptor states).1
We have investigated the molecular wire behaviour in a series of π-conjugated systems employing two different approaches: at the macroscopic level, studying the molecular wire behaviour in Donor-Bridge-Acceptor (D−B−A) systems, and at the molecular level, by studying the conductance of a single molecule connected to two electrodes though different techniques.
Donor-Bridge-Acceptor (D−B−A) systems
The direct excitation by means of an external stimuli (e.g. photoexcitation) allows studying the molecular-wire behavior. This is characterised by the attenuation factor (ß), which is obtained by measuring the charge transfer kinetics of the different D−B−A systems. Exceptionally small ß values and strong electronic donor-acceptor coupling (V) are responsible for the nanowire behaviour, assisting charge-transfer processes that exhibit a rather weak dependence on distance. ß is not purely a bridge parameter, and different values of ß can be obtained for the same oligomer connecting different donors and acceptors.
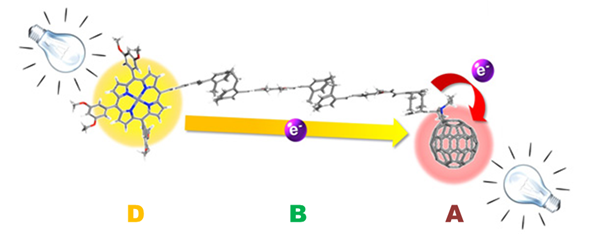
Schematic representation of a D−B−A system (ZnP-pCp-oPPV-C60 conjugates.
In our group, we have systematically employed fullerene as A and exTTF or porphyrin molecules as the electron D moieties. As for the bridges (B), a wide variety of π-conjugated systems has been evaluated, including oligophenylenevinylene (oPPV),2 oligophenyleneethynylene (oPPE),3 oligofluorene (oFL),4 oligovinylfluorene (oFV),5 oligoparacyclophane-oligophenylenevinylene (pCp-oPPV),6 and m-phenyleneethynylene.7
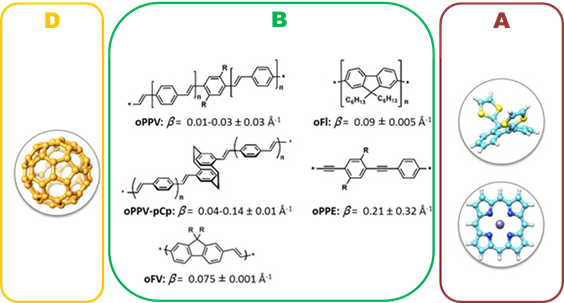
Attenuation factors (ß) for some of the Donor-Bridge-C60 systems studied.
Molecular switches
Another use a molecular wire may provide when suited with the appropriated motif is its use as switche. We have prepared molecular conjugates in which exTTF (D) and a fullerene derivative (A) are connected through a dynamic bridge of photoswitchable dithienylethenes. Exposure to an external source of light, promotes structural changes into the conjugated bridge connecting both D and A, allowing the external control of the electronic communication between the D and A units.8
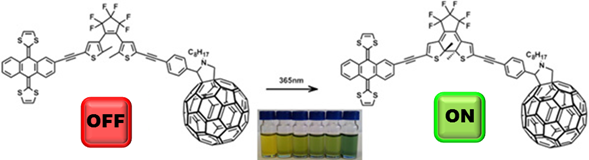
Open and closed structures (Off and On modes) of exTTF-DTE-C60.
Single molecule conductance
The direct study of the electron-transport properties through a single molecule has recently become real thanks to a series of novel techniques. Mechanically controllable break junctions (MCBJ) or scanning tunneling microscopy (STM) techniques allow measuring the conductance of single molecules sandwiched in between two metal contacts.
For this purpose, we have prepared TTF-based molecular wires endowed with thioacetyl end groups for their attachment to gold surfaces. Charge transport measurements carried out by MCBJ technique showed interesting electrostatically adjustable conductance properties, emphasising the potential of TTF derivatives to perform as molecular wires. 9
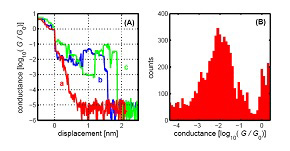
Tetrathiafulvalene-based molecular nanowire. (A) Conductance traces for an Au–Au junction (trace a) and two molecular junctions comprising one or a few molecules (traces b and c). (B) Conductance histogram constructed from the conductance traces that show evidence of trapped molecules.
A new and straightforward protocol allowed us for unambiguously isolating a single organic molecule on a metal surface and wiring it inside a nanojunction under ambient conditions.10 Employment of C60 terminal groups acting as alligator clips between the tip and the surface granted the formation of a junction with just one molecule.
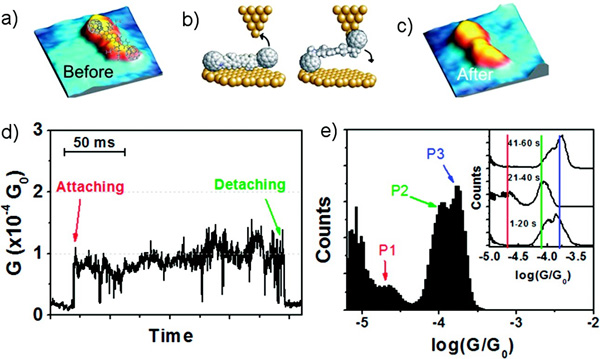
(a) 4 × 4 nm STM image of a single molecule of C60–Fl2–C60 before recording the conductance. (b) Schematic view of the molecule attaching to the tip and the surface. (c) STM image recorded after 60 s of recording the conductance. (d) Example conductance trace recorded with the tip positioned above the molecule and the feedback loop disabled. (e) Total histogram of the data recorded after disabling the feedback for 60 s.
More recently, we have investigated the junction formation with a fullerene dumbbell by pressing the STM tip onto one of the C60 anchor groups to lift it, leaving it dangling from the tip and with the second C60 unit free to bind the surface. Two main conductance states were observed, a high conductance state close to 10-2 G0 and a lower state close to 10-4 G0. Theoretical calculations suggest that the high conductance state corresponds to current injection directly into the fluorene bridge, while the reduction in the conductance is probably due to the trapping of a molecule of solvent or a solvent monolayer between the surface and one C60 group.11,12
Graphene nano-electrodes
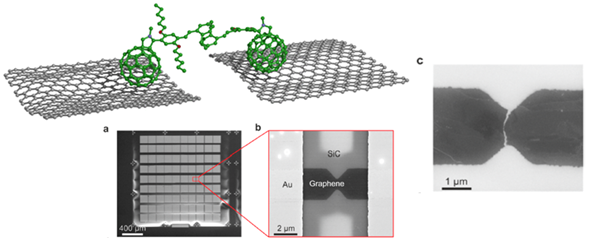
Schematics of the pCp-oPPV dumbbell wire. a,b &c show different augmentations of the graphene based electrodes.
On the way to ultraflat single-molecule junctions with transparent electrodes, we elaborated a fabrication scheme based on the use of epitaxial graphene nano-electrodes. By using a pCp-oPPV molecular-wire with fullerene anchoring groups, stable electrical properties could be recorded. Electrical measurements showed that single-molecule junctions with graphene electrodes displayed strikingly similar results to the ones obtained with gold electrodes. Further demonstrating that the molecules were almost insensitive to the electrode nature.13
Anchoring groups
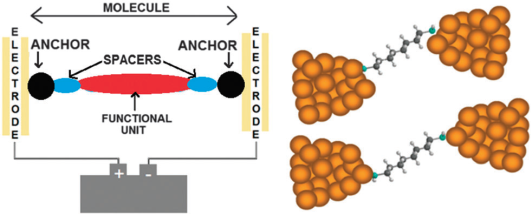
(Left) A schematic representation of a single molecule junction highlighting the key components, namely the anchor groups, a functional unit, spacers (alkyl chains for example) and the enclosing electrodes. (Right) More realistic representations of a junction showing different atomic configurations of the binding to the electrodes.
When a molecule is wired between a pair of electrodes, the two points of contact are determined by the chemical anchoring groups, located at the ends of the molecule. At this point, when a bias is applied, electrons are channelled from a metallic environment through an extremely narrow constriction, essentially a single atom, into the molecule. The fact that this is such an abrupt change in the electron pathway makes the nature of the chemical anchoring groups critically important regarding the propagation of electrons from the electrode across the molecule. A delicate interplay of phenomena can occur when a molecule binds to the electrodes, which can produce profound differences in conductance properties depending on the anchoring group. This makes answering the question “what is the best anchoring group for single molecule studies” far from straight forward.14
References
1. D. M. Guldi, B. Illescas, C. Atienza, M. Wielopolski, N. Martín, Chem. Soc. Rev. 2009, 38, 1587.
2. (a) F. Giacalone, J. L. Segura, N. Martín, D. M. Guldi, J. Am. Chem. Soc. 2004, 126, 5340; (b) F. Giacalone, J. L. Segura, N. Martín, J. Ramey, D. M. Guldi, Chem. Eur. J. 2005, 11, 4819; (c) G. de la Torre, F. Giacalone, J. L. Segura, N. Martín, D. M. Guldi, Chem. Eur. J. 2005, 11 1267.
3. (a) C. Atienza, N. Martín, M. Wielopolski, N. Haworth, T. Clark, D. M. Guldi, Chem Commun. 2006, 3202; (b) M. Wielopolski, C. Atienza, T. Clark, D. M. Guldi, N. Martín, Chem. Eur. J. 2008, 14, 6379.
4. C. Atienza-Castellanos, M. Wielopolski, D. M. Guldi, C. van der Pol, M. R. Bryce, S. Filippone, N. Martín, Chem. Commun. 2007, 5164.
5. M. Wielopolski, J. Santos, B. M. Illescas, A. Ortiz, B. Insuasty, T. Bauer, T. Clark, D. M. Guldi, N. Martín, Energy Environ. Sci. 2011, 4, 765.
6. (a) A. Molina-Ontoria, M. Wielopolski, J. Gebhardt, A. Goloumis, T. Clark, D. M. Guldi, N. Martín, J. Am. Chem. Soc. 2011, 133, 2370; (b) M. Wielopolski, A. Molina-Ontoria, C. Schubert, J. T. Margraf, E. Krokos, J. Kirschner, A. Goloumis, T. Clark, D. M. Guldi, N. Martín, J. Am. Chem. Soc. 2013, 135, 10372.
7. A. Molina-Ontoria, G. Fernández, M. Wielopolski, C. Atienza, L. Sánchez, A. Gouloumis, T. Clark, N. Martín, D. M. Guldi, J. Am. Chem. Soc. 2009, 131, 12218.
8. S. Castellanos, A. A. Vieira, B. M. Illescas, V. Sacchetti, C. Schubert, J. Moreno, D. M. Guldi, S. Hecht, N. Martín, Angew. Chem. Int. Ed. 2013, 52, 13985.
9. F. Giacalone, M.A. Herranz, N. Agrait, L. Grüter, M.T. Gónzalez, M. Calame, C. Schönenberger, N. Martín, Chem. Commun. 2007, 4854.
10. E. Leary, M. T. González, C. van der Pol, M. R. Bryce, S. Filippone, N. Martín, G. Rubio-Bollinger, N. Agraït, Nano Lett. 2011, 11, 2236.
11. K. Gillemot, C. Evangeli, E. Leary, A. La Rosa, M. T. González, S. Filippone, I. Grace, G. Rubio-Bollinger, J. Ferrer, N. Martín, C. J. Lambert, N. Agraït, Small 2013, 9, 3812.
12. P. Moreno-García, A. La Rosa, V. Kolivoska, D. Bermejo, W. Hong, K. Yoshida, M. Baghernejad, S. Filippone, P. Broekmann, T. Wandlowski, N. Martín, J. Am. Chem. Soc. 2015, 137, 2318.
13. K. Ullmann, P. B. Coto, S. Leitherer, A. Molina-Ontoria, N. Martín, M. Thoss, H. B. Weber, Nano Lett. 2015, 15, 3512.
14. E. Leary, A. La Rosa, M. T. González, G. Rubio-Bollinger, N. Agraït, N. Martín, Chem. Soc. Rev. 2015, 44, 920.